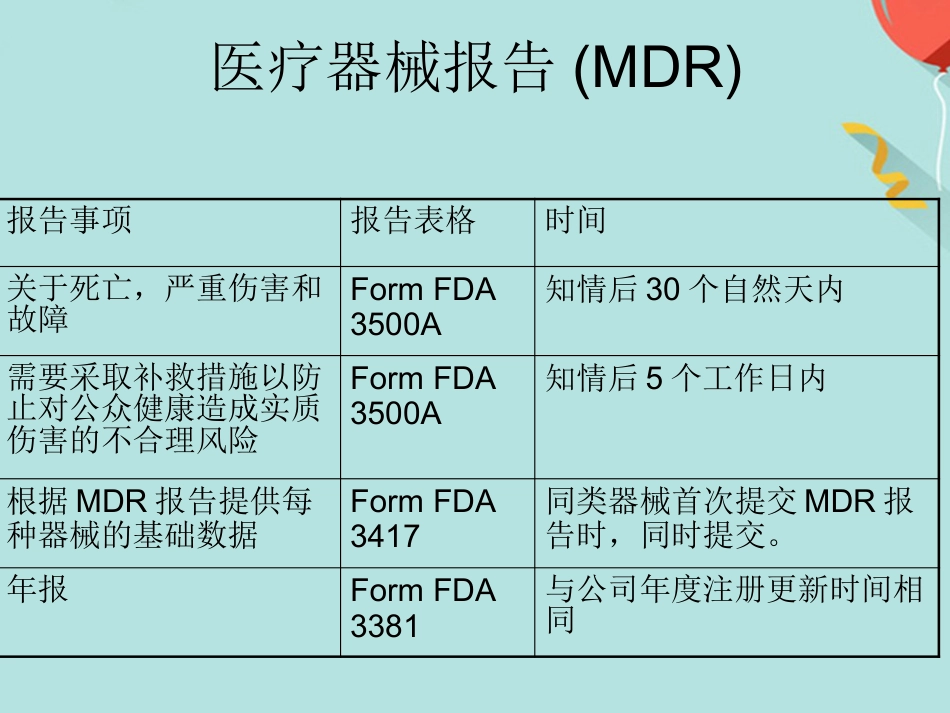
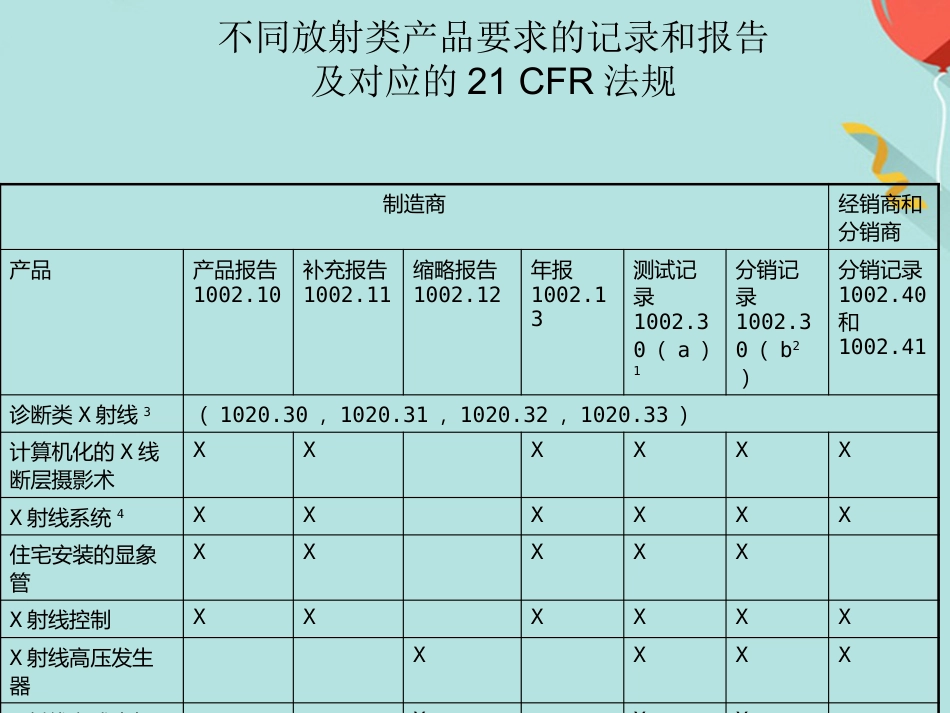
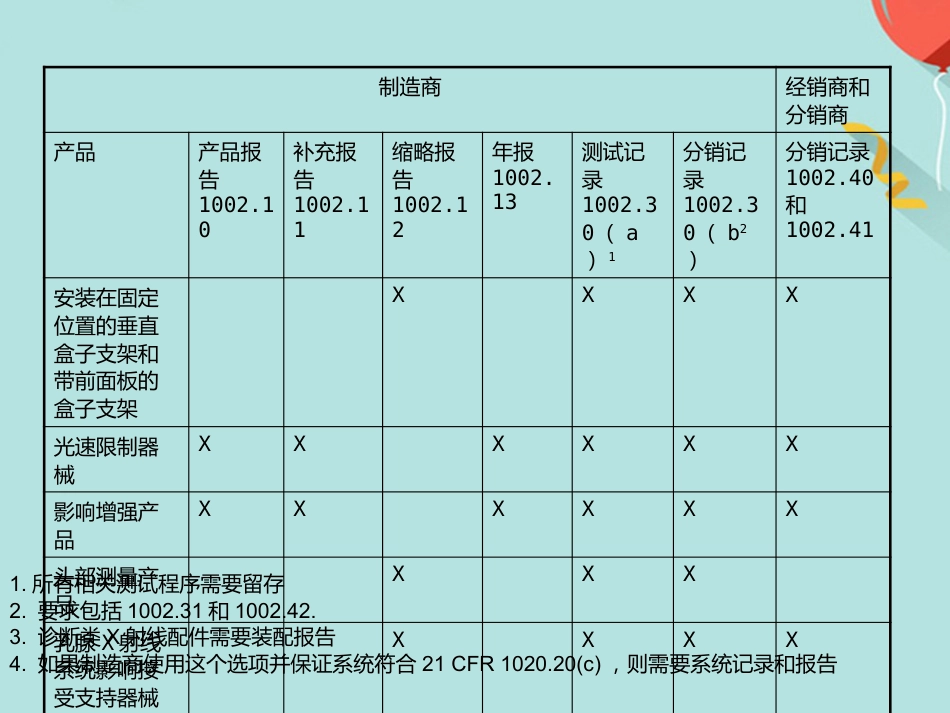
FDA 法规讲座1. 法规综述2. CDRH 管辖的产品3. 器械分类4. 产品的合法销售5. 企业注册6. 产品列示7. 510(k) 报告FDA 法规简介•介绍•入市前许可 (PMA)•质量体系法规(QSR )•标识要求•医疗器械报告(MDR )•医疗器械使用者费用和2002 现代化法案(MDUFMA )介绍 与医疗器械相关的法规要求:除非获得豁免,否则需要入市前通知(510(k)) 或入市前许可(PMA)企业注册( 包括FDA-2891 表格)医疗器械列示(FDA-2892 表格)质量系统(QS) 法规标识要求和医疗器械报告(MDR)- 售后监督体系入市前许可(PMA )• PMA 适用于高风险性的三类医疗器械或无法通过510(k) 途径找到合适实质性等同器械的一类和二类产品。• 2003 财政年度开始收费研究用的器械豁免(IDE )• IDE -在人体上应用未获得许可的医疗器械进行临床试验。• 高风险性的产品进行临床研究必须由FDA 和审查委员会 (Institutional Review Board) 同时批准。• 非高风险性产品进行临床研究必须由IRB 批准。IRB- 由某个机构正式授权,可以进行与人类相关生物医学研究的审核,初始化批准以及操作过程审核的部门。质量体系法规(QSR )/ 良好生产规范(GMP )• QSR 包括运用于设施的方法以及运用于医疗器械设计,采购,生产,包装,标识,储存,安装以及服务的控制。• FDA 检查的生产厂家必须满足QSR 要求。• 医疗器械的设计和发展必须满足质量体系法规中的设计控制要求。标识• 标识包括医疗器械上的标签以及所有与该器械有关的描述性和信息性的文字。说明书,包装标签,广告等都属于标示。医疗器械报告(MDR)• 必须汇报的事故包括:器械导致或与死亡或严重伤害相关的事故• MDR 法规是FDA 和生产商用于识别和监控重大负面事件包括医疗器械的一种机制。• MDR 的目的-及时的发现和处理问题。医疗器械报告(MDR)报告事项报告表格时间关于死亡,严重伤害和故障Form FDA 3500A知情后30 个自然天内需要采取补救措施以防止对公众健康造成实质伤害的不合理风险Form FDA 3500A知情后5 个工作日内根据MDR 报告提供每种器械的基础数据Form FDA 3417同类器械首次提交MDR 报告时,同时提交。年报Form FDA 3381与公司年度注册更新时间相同医疗器械使用者费用和2002 现代化法案(MDUFMA )• 2002 年10 月26 日医疗器械使用者费和MDUFMA 立法。• MDUFMA 的三个显著规定:入市前审核费用企业检查可以由第三方进行关于一次性...