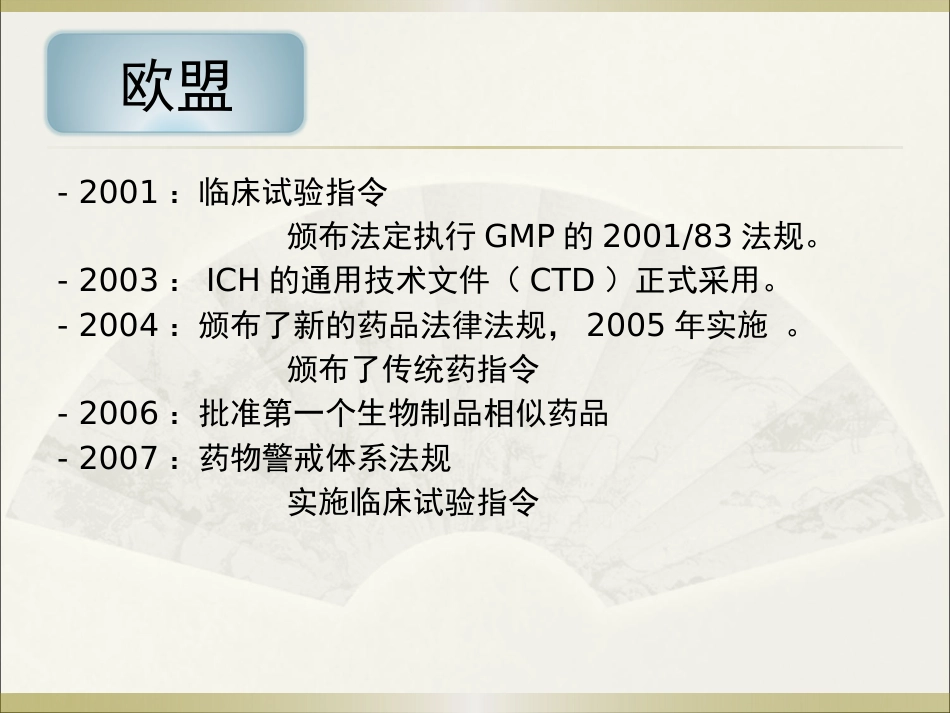
欧洲背景欧洲法律法规体系欧洲上市许可体系欧洲监督检查体系欧洲监管当局体系欧洲药典会认证项目欧洲药典会国际协调欧盟欧洲背景1949 - 欧洲理事会, The Council of Europe, 47 个国家, 近8 亿人口,政治共同体,旨在社会发展。1952 - 欧洲煤钢共同体1958 - 改为欧洲共同体(1957 年罗马协议)1965 - 称共同体1993 – 欧盟, The European Union , 25 个国家,共同体统合在欧洲联盟下, 贸易实体转为经济 和政治联盟,单一市场, 旨在人员、货物自由流动。药品是单一市场的目标之一。涵盖5 亿人口。欧盟欧洲药品法律法规体系 - 1965 :颁布了第一个药品监管指令,制定了基本原则 - 1975 :颁布了第一个关于药品检验和执行标准的指令 - 1985 :“ 1992 单一市场”计划启动 - 1993 :理事会法规2309/93EEC 和其他法规,包括欧洲药品上市许可新程序和监督法规 - 1995 :正式实施药品集中批程序 - 1998 :强制实施互认程序 - 2000 :颁布孤儿药法欧盟- 2001 :临床试验指令 颁布法定执行GMP 的2001/83 法规。 - 2003 :ICH 的通用技术文件(CTD )正式采用。- 2004 :颁布了新的药品法律法规,2005 年实施 。 颁布了传统药指令- 2006 :批准第一个生物制品相似药品 - 2007 :药物警戒体系法规 实施临床试验指令欧盟药品上市许可体系 - 集中审批程序(CP ) - 双方互认程序(MRP ) - 非集中程序(DP ) - 国家审批程序(NP )欧盟新药分类:Part A: 生物技术产品 Part B: 新活性物质 - 重组DNA 产品 - 重大创新- 基因表达生物活性蛋白 - 重大治疗价值- 混合和单克隆抗体观 - 来自人体血液/ 血浆新产品 - 新活性物质欧盟强制集中审批:A 类产品,即生物技术产品 2005 年11 月起,增加强制集中审批的新活性物质品种:用于治疗糖尿病癌症、获得性免疫缺陷病(HIV ),神经失调(海兹默病等)孤儿药 (危机生命的或严重疾病,其发病率低于万分之五,10 年市场独占权,集中审批、减低集中审批程序所要交费标准,EU 支持研究开发经费支持)欧盟2008 年5 月起,增加强制许可品种:自主免疫疾病和其他免疫缺陷病毒性疾病其他根据欧盟提出的经理事会批准的扩大品种Part B: 新活性物质 (自愿)欧盟双方互认程序 - 大部分普通药品,销往两个以上成员国国家。 (一国批准后,可向另一...